
Diagnosis of mitochondrial diseases
“Development of a personalized medicine approach to improve diagnosis of mitochondrial diseases” is Justine Labory’s PhD research topic. It was funded by the PIA MITOMICS project and currently carried out at IRCAN under the supervision of Prof. Sylvie Bannwarth and with the support of MSI and in particular Dr. Silvia Bottini.
Summary
Mitochondrial diseases (MD) are rare pathologies caused by a deficiency in the mitochondrial respiratory chain (MRC), which supplies energy to cells through oxidative phosphorylation. They are the most common metabolic disorders, with an estimated incidence of 1/5,000 births. MD are extremely heterogeneous, both clinically and genetically, with a wide age range and very different symptoms, making them very difficult to diagnose.
MD are caused by variants in genes involved in mitochondrial biogenesis. Although mitochondria have their own genome, over 1,000 nuclear genes are required for MRC function. Consequently, MD can be caused by pathogenic variants affecting either mtDNA or nuclear genes. Diagnosis requires identification of the gene responsible.
Today, over 400 nuclear genes are implicated in MD, and the list continues to grow. Despite the development of high-throughput sequencing (NGS), more than one patient in two remains at a diagnostic impasse, as the gene responsible remains unknown.
The advent of whole exome sequencing (WES) and whole genome sequencing (WGS) has considerably accelerated the identification of variants in previously unknown disease genes. Nevertheless, their success rate in detecting the responsible variants is only between 25 and 50%. Many of the variants detected by WES remain variants of unknown significance (VUS), or are omitted due to the impossibility of prioritizing them. In addition, variants present in non-coding regions cannot be identified by WES. WGS detects variants in both coding and non-coding regions, but presents a significant (and currently unresolved) data interpretation challenge for the more than 3 million variants per sample.
Recent studies have demonstrated the value of RNA sequencing (RNAseq) in selecting candidate genes with altered or monoallelic expression, and in identifying deep intronic variants affecting splicing, when used in combination with WES. For MD, the use of RNAseq in 48 undiagnosed patients after WES increased the success rate by 10%.
In this context, the development of new bioinformatics approaches is essential to resolve the diagnostic impasse and improve our knowledge of MD.
Partners
- Université Angers
- Université Côte d'Azur
- IRCAN
- Inria Center at Université Côte d'Azur
- Université de Nantes
- Angers University Hospital
- Nice University Hospital
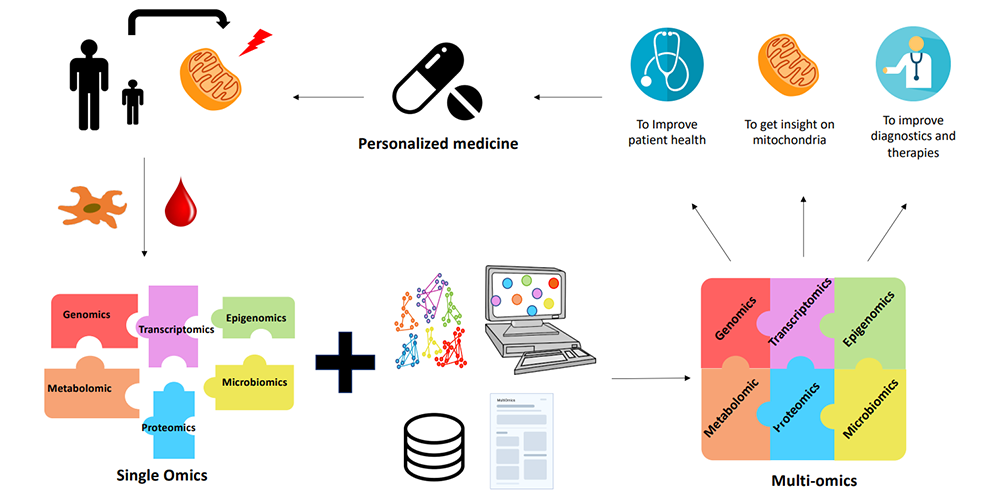
Image: The contribution of multiomics to MD. Blood or tissue samples (skin or muscle biopsy) are taken from MD patients. Fibroblasts or myotubes are prepared and cultured from tissue biopsies. Different omics analyses are performed separately to identify the gene(s) responsible for the pathology. However, these analyses are not always conclusive. Despite the validity of the simple omics approach, the complexity of the MD limits the success of the diagnosis. This is where multi-omics comes in. Thanks to bioinformatics and the development of algorithms, it is now possible to integrate data from several different omics analyses, and obtain better results. Multi-omics, combined with databases and literature, provide a better understanding of MD. As a result, diagnosis is improved and personalized treatment can be proposed for each patient.